You must login before you can run this tool.






Category
Published on
Abstract
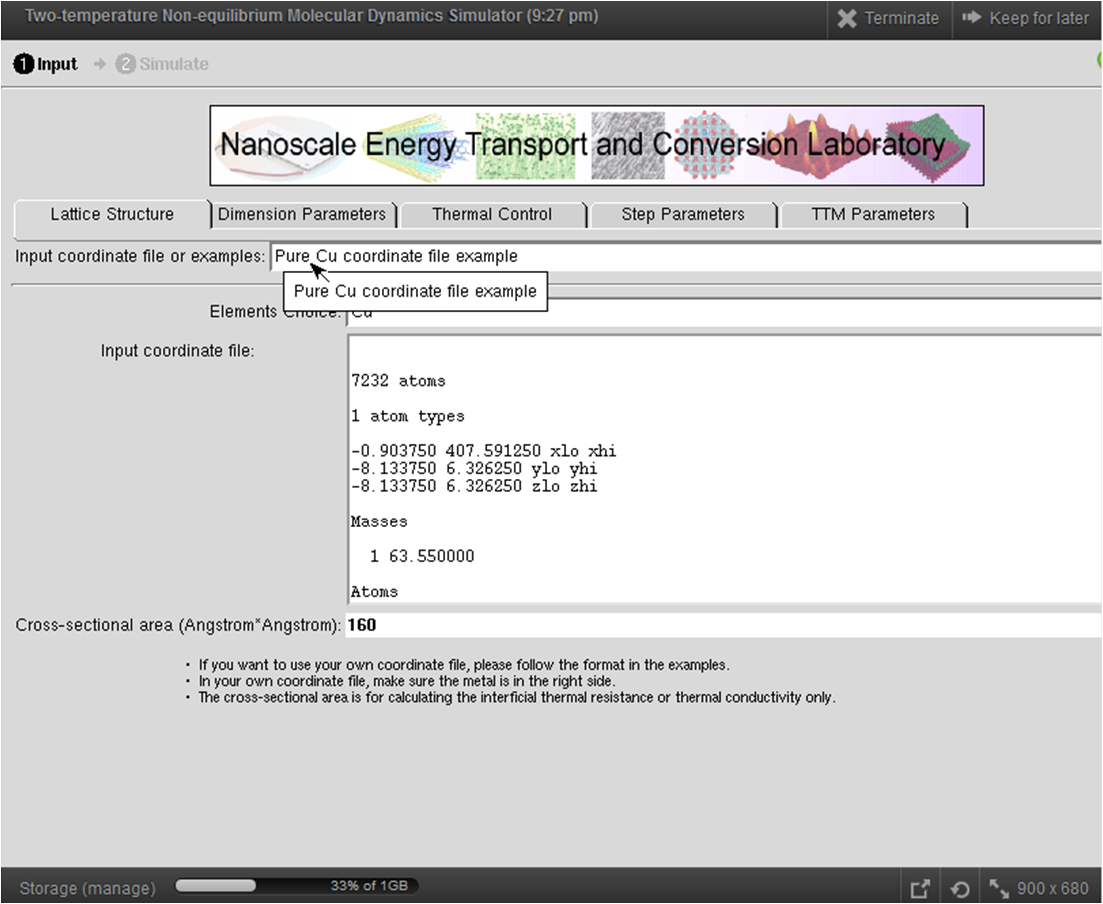
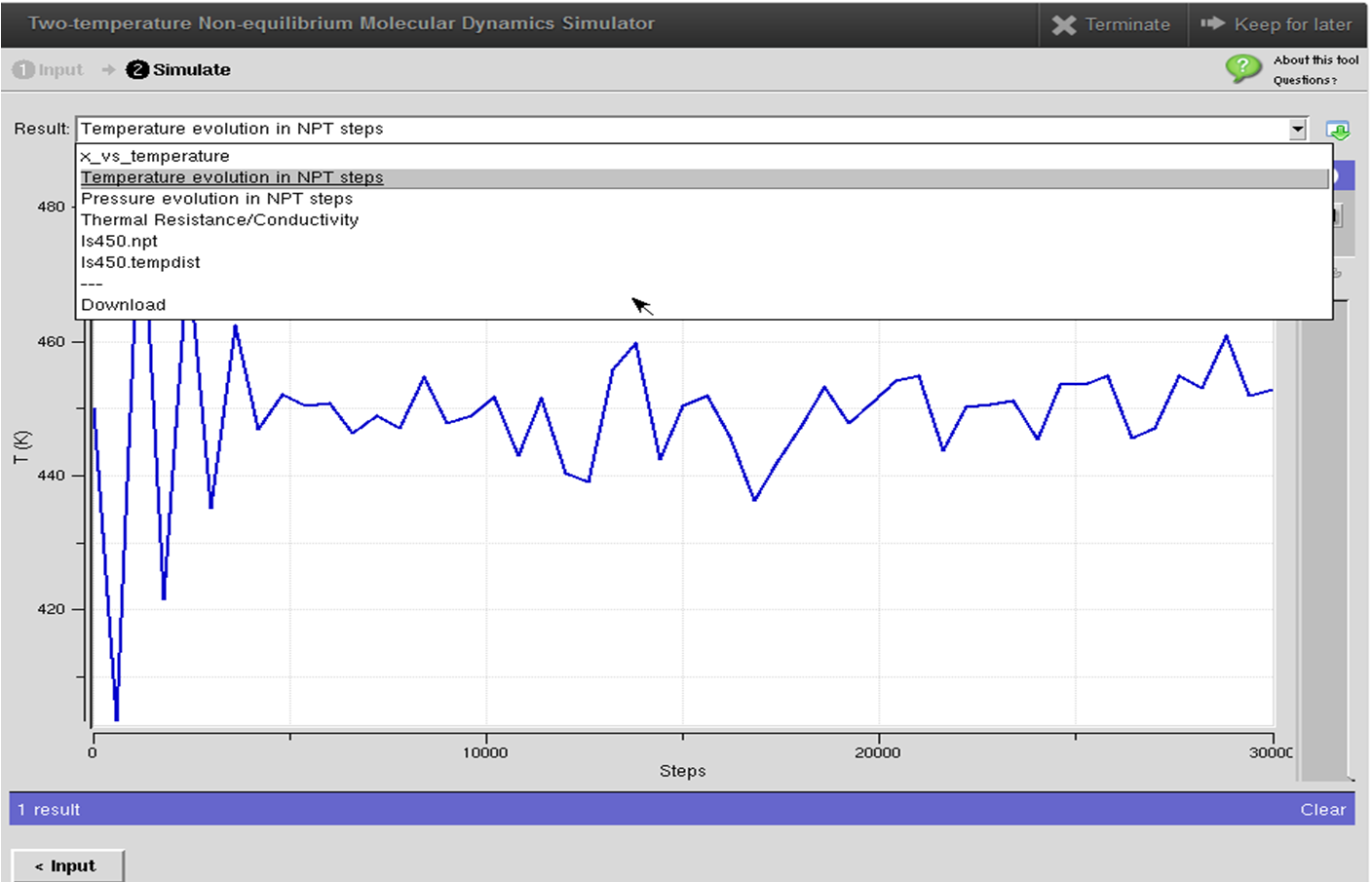
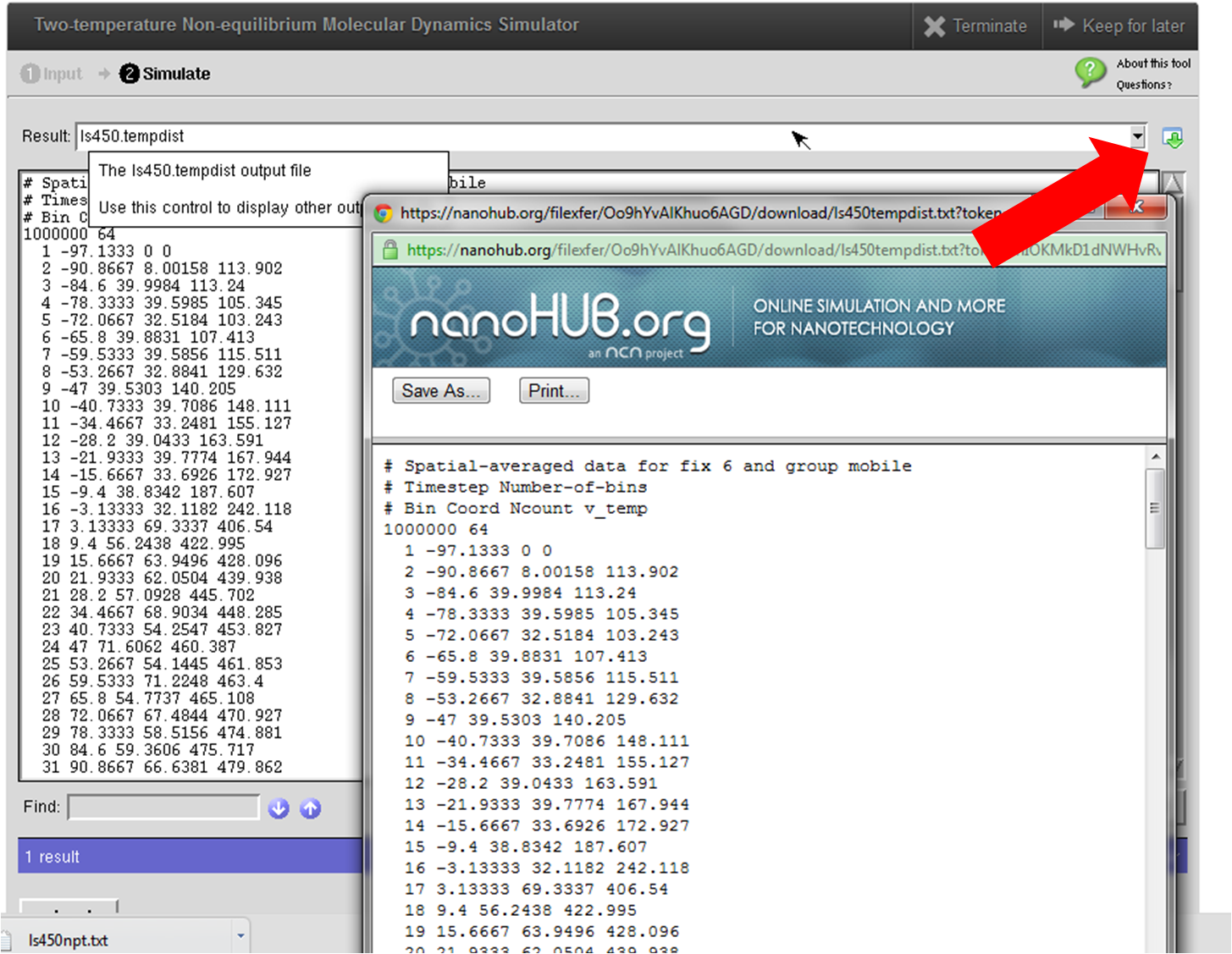
This tool combines the conventional molecular dynamics (LAMMPS) for phonons with the two-temperature model (TTM) to incorporate the electronic thermal transport channel. It can thus model electron-phonon coupled thermal transport across interfaces between metals and dielectrics. A detailed user's guide of this tool can be found in this page. This tool contains several example structures, e.g., Si/Cu, CNT/Cu and pure Cu, which allows first-time users to get familiar with the two-temperature nonequilibrium molecular dynamics simulation (TTM-MD) more quickly. The structures used as examples are quite small so as to achieve faster simulation and are good for demonstration purpose only. To obtain publication level results, we recommend the users to simulate several structures of different sizes to do a systematical study on the size effect and, hence, eliminate the size effect. The users can switch the molecular dynamics simulation mode between two-temperature nonequilibrium molecular dyanmics (electrons and phonons) and conventional MD (only phonons) by checking/unchecking the corresponding box in the "TTM Parameters" section of the tool. By comparing the obtained temperature profiles and the thermal boundary resistance, the users can make a good sense of the importance of the electronic effect to interfacial thermal transport across metal-nonmetal interfaces. Again, we highly recommend reading the User's Guide before doing higher-level simulations.
Powered by
the LAMMPS Molecular Dynamics Simulator and RAPPTURE
Credits
Yan Wang, School of Mechanical Engineering and the Birck Nanotechnology Center, Purdue University, West Lafayette, IN 47907; Xin Jin, School of Electrical and Computer Engineering, Purdue University, West Lafayette, IN 47907; Xiulin Ruan, School of Mechanical Engineering and the Birck Nanotechnology Center, Purdue University, West Lafayette, IN 47907; Ajit K. Roy, Materials and Manufacturing Directorate, Air Force Research Laboratory, Wright Patterson Air Force Base, Dayton, Ohio 45433, USA; The authors acknowledge Dr. Steven Clark from the Nanohub team for his help in publishing the first version of this tool.
Sponsored by
The Air Force Office of Scientific Research (AFOSR). The NSF CAREER Award. Purdue Network for Computational Nanotechnology (NCN). Purdue Summer Undergraduate Research Fellowship Program (SURF).
Publications
Y. Wang, X.L. Ruan, and A.K. Roy, "Two-temperature nonequilibrium molecular dynamics simulation of thermal transport across metal-nonmetal interfaces", Phys. Rev. B 85, 205311, 2012. http://prb.aps.org/abstract/PRB/v85/i20/e205311
Cite this work
Researchers should cite this work as follows: