Ab initio electronic structure simulation tools
ABINIT
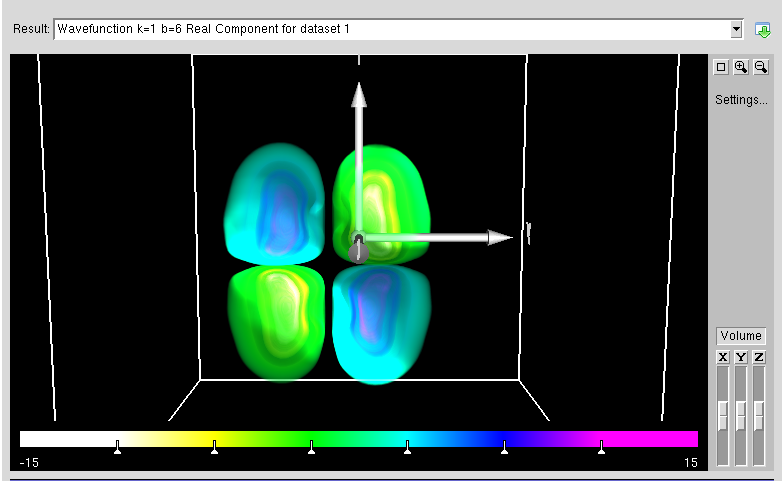
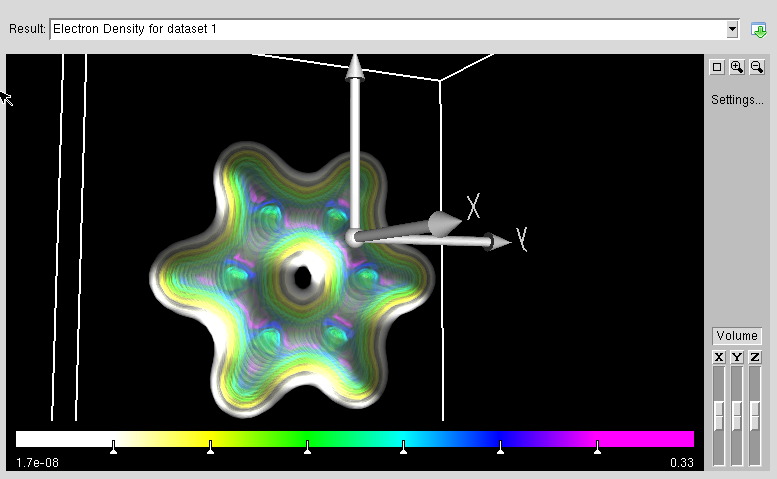
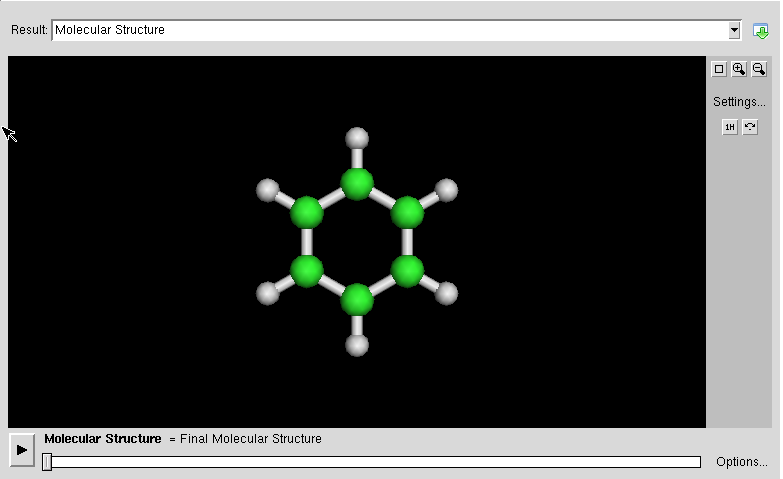
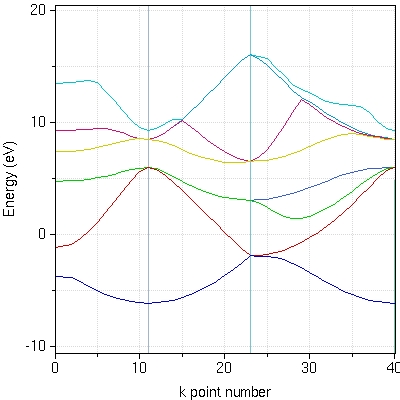
- :ABINIT is a package whose main program allows to find the total energy, charge density and electronic structure of systems made of electrons and nuclei (molecules and periodic solids) within Density Functional Theory, using pseudopotentials and a planewave basis. ABINIT also includes options to optimize the geometry according to the DFT forces and stresses, or to perform molecular dynamics simulation using these forces, or to generate dynamical matrices, Born effective charges, and dielectric tensors. In addition to the main ABINIT code, different utility programs are provided.
- :First Time User Guide for ABINIT on nanoHUB.org
- :Official ABINIT site
- :ABINIT New User Guide
SIESTA
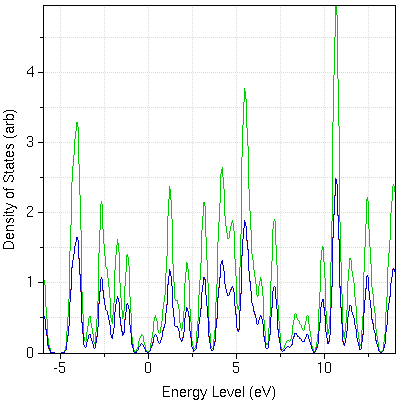
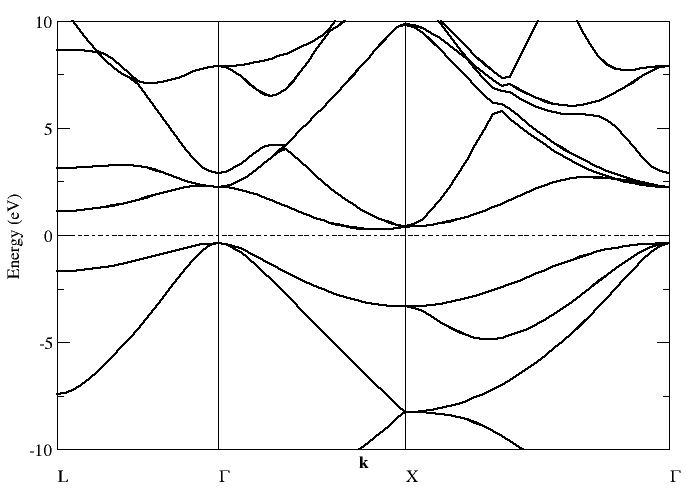
- : Siesta is a fast electronic structure program that uses a local basis. It can produce band structures and minimum energy geometries, and can perform first principles molecular dynamics calculations.
- :siesta Website
nanoMATERIALS SeqQuest DFT
- :powered by
SeqQuest - :This tool provides access to the density functional theory code
SeqQuestvia nanoHUB.org.SeqQuestis being developed at Sandia National Laboratories by Dr. Peter A. Schultz of the Multiscale Dynamic Materials Modeling Department and collaborators. UsingSeqQuestthe tool enables users to calculate the total energy, atomic forces and stress for molecules, wires, slabs and bulk systems. - :seqQuest webpage
QC-Lab
- :powered by GAMESS
- :QC-Lab – Quantum Chemistry Lab. It provides Ab Initio and DFT molecular and electronic structure calculations of small molecules.
- :GAMESS Website
StrainBands
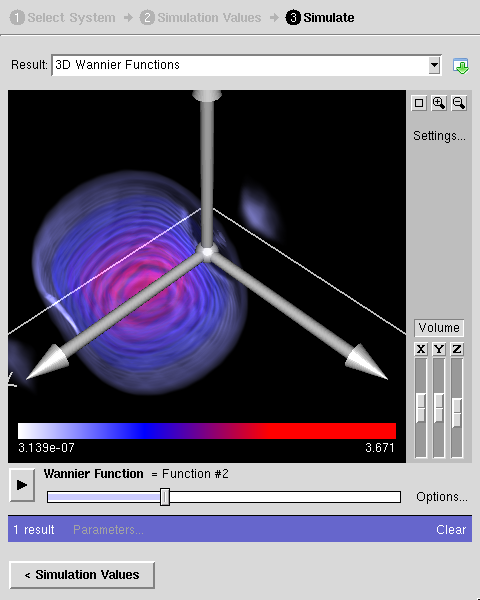
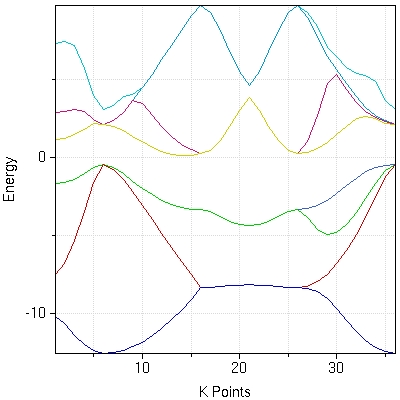
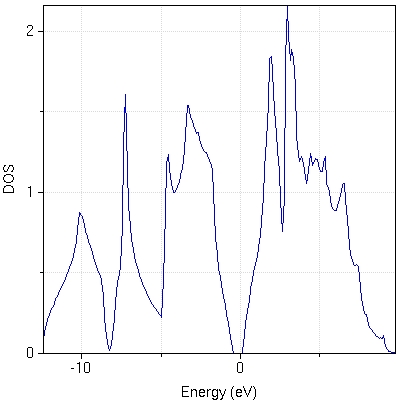
- :powered by: Electronic structure calculations performed by [PWscf and Quantum-Espresso v3.2.2]. Maximally-localized Wannier functions calculated by [Wannier90 v1.0.2].
- :This tool can be used to explore the influence of strain on first-principles bandstructures of semiconductors.
DFT calculations with Quantum ESPRESSO
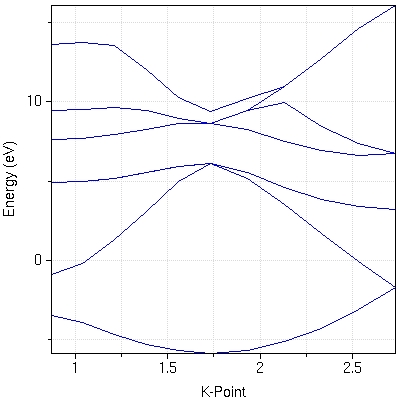
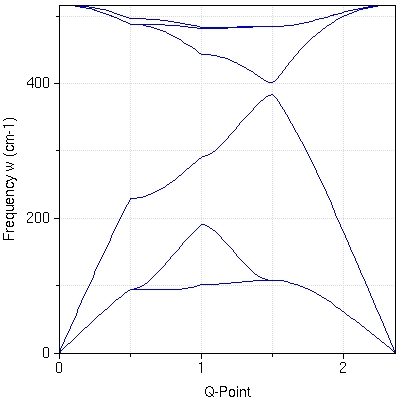
- :This tool enables nanoHUB users to run quantum ESPRESSO, a powerful electronic structure code. Users can perform total energy calculations, energy minimization to predict structures, obtain the Kohn-Sham band structure of periodic systems as well as phonons.
QWalk Quantum Monte Carlo Tutorial
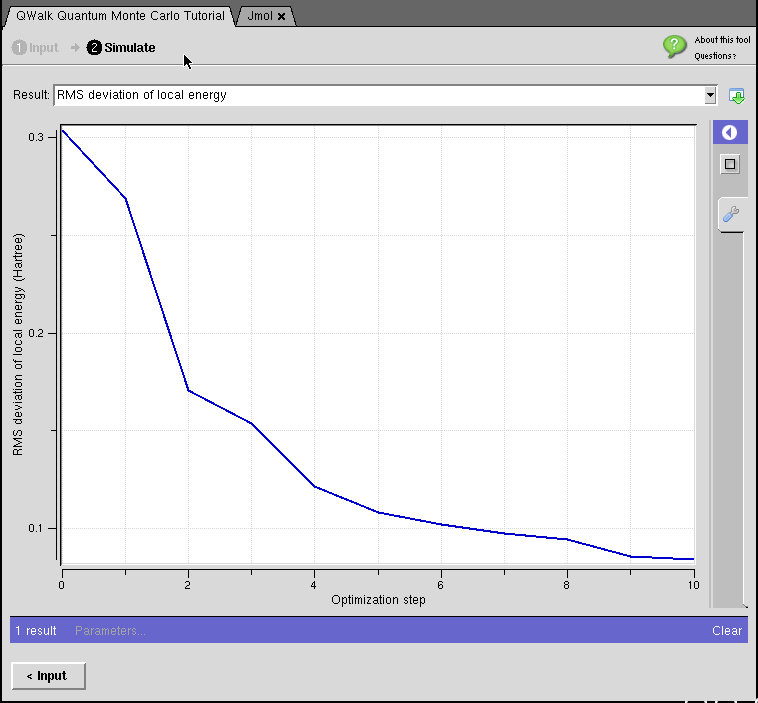
- : Quantum Monte Carlo methods solve the Schrodinger equation for many electrons to high accuracy—exactly in some cases. In most implementations, it also has favorable scaling with system size, approximately the same as mean-field theories like density functional theory, although with a larger prefactor. This allows us to obtain accurate ground and excited state energies for realistic chemical systems. Quantities such as binding energies, reaction barriers, and band gaps are accurately simulated using QMC methods.
- :qwalk Website
Molecular dynamics simulations Tools
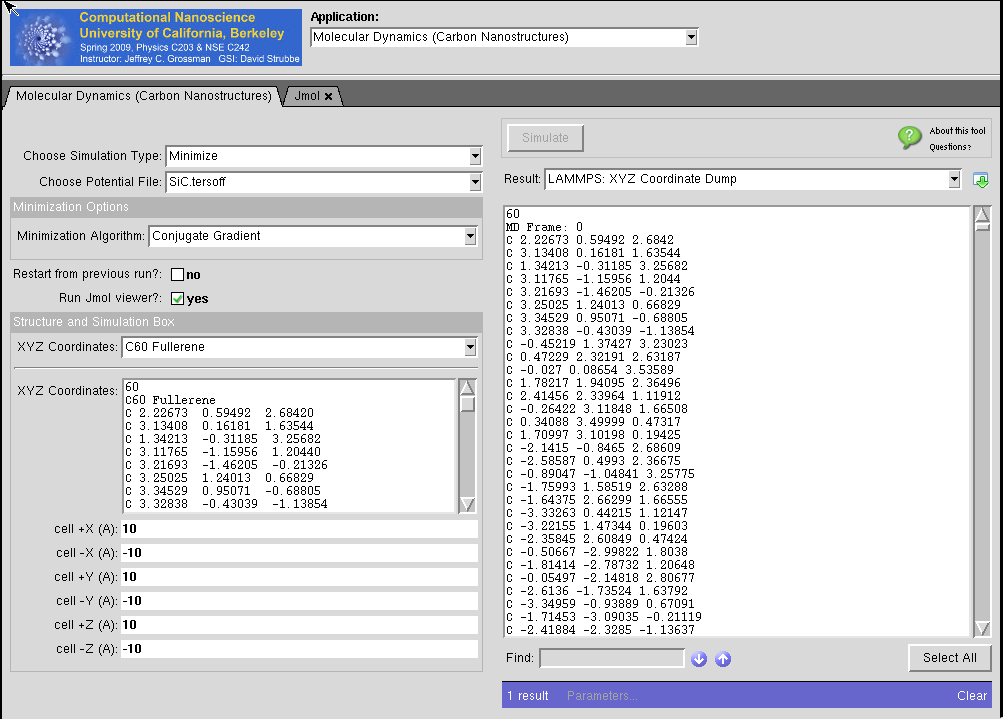
nano-Materials Simulation Toolkit
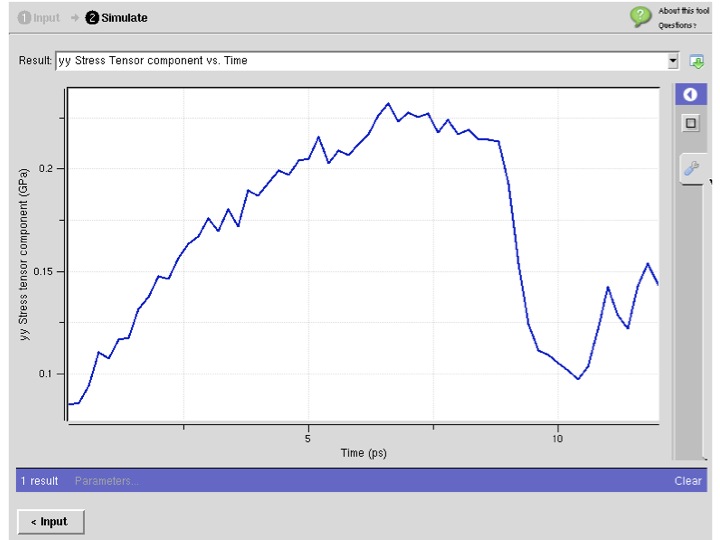
- : powered by Strachan Group MD
- :The nanoMATERIALS simulation toolkit enables users to perform molecular dynamics simulations of materials using a variety of force fields as well as electronic structure calculations.
- :Atomic Picture of Plastic Deformation in Metals
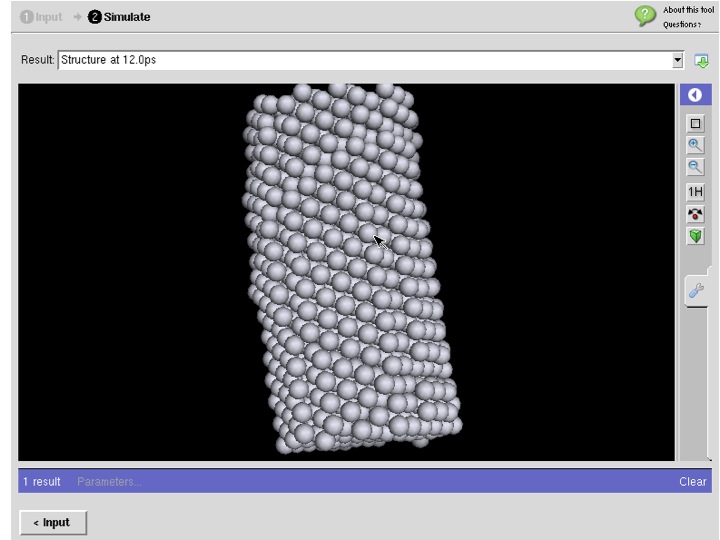
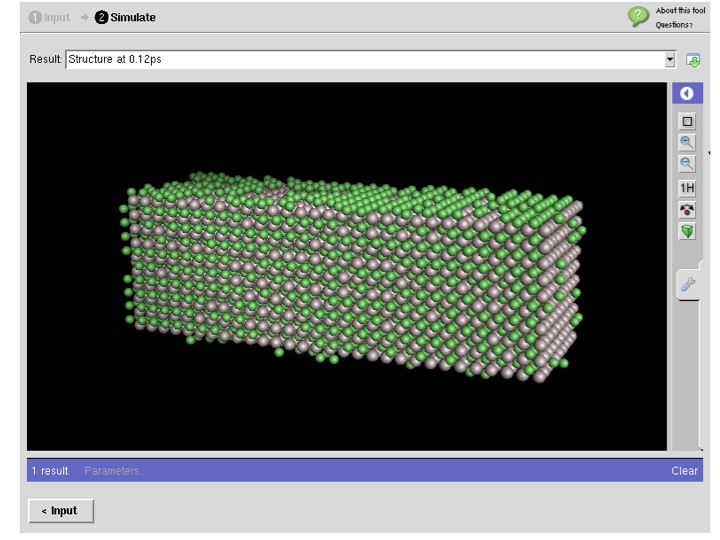
LAMMPS for Carbon Nanostructures in MIT Atomic-Scale Modeling Toolkit

- : powered by LAMMPS
- :This tool is part of MIT Atomic-Scale Modeling Toolkit which serves Overview of Computational Nanoscience: a UC Berkeley Course
- :LAMMPS Website
Lennard-Jones Potential with LAMMPS in MIT Atomic-Scale Modeling Toolkit
- : powered by LAMMPS
- :This tool is part of MIT Atomic-Scale Modeling Toolkit which serves Overview of Computational Nanoscience: a UC Berkeley Course
- :LAMMPS Website
Educational Material
Bonding and Bandstructure in Silicon is a Learning Module with Introductory Lectures, Tutorials and a Online-simulation Lab Assignment to explore how electronic bands form in Silicon and other materials. Audience: undegraduate and graduate students as well as instructors interested in electronic properties of materials.
Molecular dynamics simulations of materials is a topics page that summarizes educational resources and tools for molecular dynamics simulations.Audience: researchers interested in atomistic materials modeling and simulation.