Materials Science

Molecular Dynamics
Use this link to find all content in nanoHUB related to LAMMPS
This link creates a "tag search" for LAMMPS. You can also click on a "LAMMPS" tag to get to the same page.
Filter the results using the menu on the right side of the results page.
LAMMPS
This tool allows users who are experienced with LAMMPS to upload a command script and data file for any system and run LAMMPS either as a serial calculation or a parallel run on a HPC system. Users can upload command scripts with pair_style kim, and the tool will automatically download and load Models from the OpenKIM repository.
Crack Propagation Lab

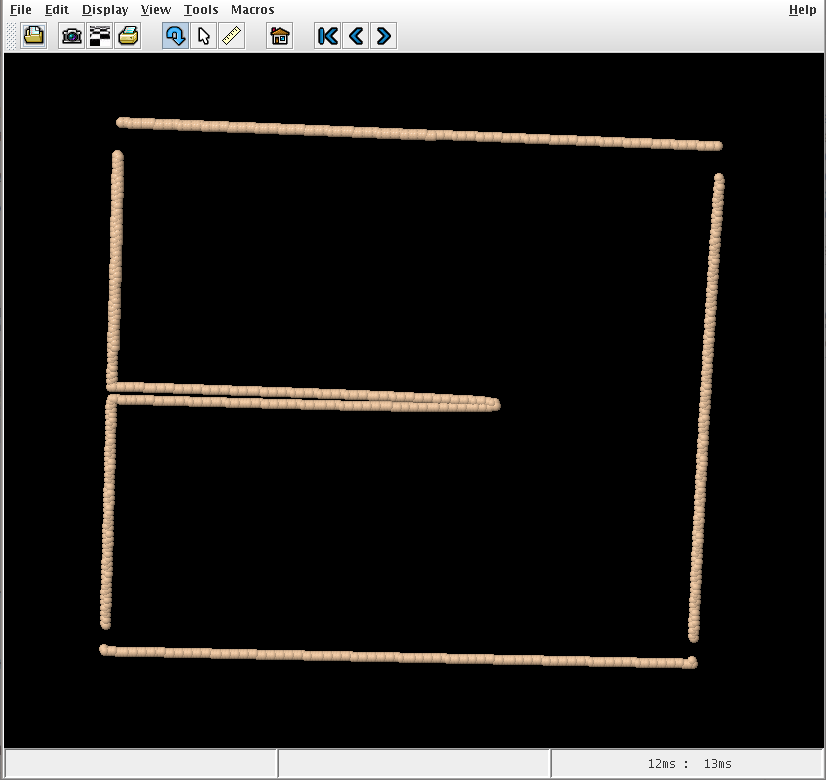
This tool models supersonic crack propagation in a 2D triangular lattice with pair interaction potentials between atoms.
Nanomaterial mechanics explorer


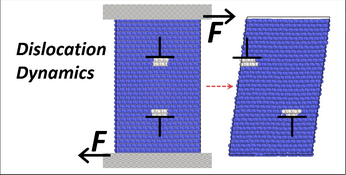
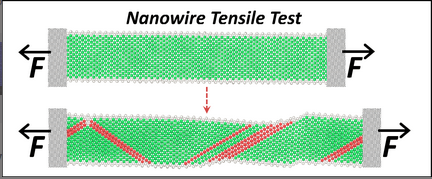
This tool is for those who would like to explore fundamental properties of materials such as dislocations, crack propagation, nanowire tensile testing, melting and the martensite transformation through atomistic Molecular Dynamics simulations. All simulations in this explorer are powered by the LAMMPS simulation code. The tool is set up for default operation with sets of preselected inputs for those who are new to MD simulation. Enabling the advanced functions allows users to define simulation parameters for their specific needs.
This set of tools will allow you to:
- visualize how dislocations either glide or nucleate in a crystal based on the applied stress direction relative to the Burgers vector, slip plane, and dislocation line.
- visualize how a nanowire deforms under uni-axial tensile loading, observe the process of yielding and necking, and simulate values of key engineering parameters such as the Young's modulus and yield stress. Visualize a defect in a Nickel(FCC) or Tantalum(BCC) that under uniaxial tension grows into a crack that will cause brittle fracture. Stress-strain curve, yield stress and yield strain are generated, and advanced options allow study of the brittle to ductile transition in BCC metals.
- visualize melting at the atomic level, and generate a radial distribution function. The effect of pressure on melting temperature can be studied.
- visualize a fast quench of two Ni-Al alloys, and identify the martensite transformation.
How to download simulation files for local use
Instructions for downloading the simulation results from nanoHUB for local use.
nano-Materials Simulation Toolkit


Powered by Strachan Group MD
The nanoMATERIALS simulation toolkit enables users to perform molecular dynamics simulations of materials using a variety of force fields as well as electronic structure calculations.
nanomaterials Simulation Toolkit Tutorial
Hands-on tutorial that will get you started with the nanomaterials simulation toolkit.
Learning Module— Atomic Picture of Plastic Deformation in Metals
This learning module describes how this simulation tool can be used to teach concepts about plastic deformation to sophomore-level MSE students.
Nanowire Tensile Deformation Lab
This tool allows the user to simulate the effects of applying tensile stress at each end of a Copper nanowire.
MIT Atomic-Scale Modeling Toolkit
The following simulations are run by the tool:
- Averages and Error Bars
- Molecular Dynamics (Lennard-Jones)
- Molecular Dynamics (Carbon Nanostructures and More)
- Monte Carlo (Hard Sphere) * Monte Carlo (Ising Model)
- Quantum Chemistry (GAMESS) * Density-Functional Theory (Quantum Espresso)
- Density-Functional Theory (SIESTA)
- Quantum Monte Carlo (QWalk)
nanoMATERIALS nanoscale heat transport
This tool will enable the users to calculate two heat transport properties: thermal conductivity and phonon relaxation time. The first one is to run thermal conductivity simulations on various Si/Ge structures by non-equilibrium MD with LAMMPS package. Pure Si/Ge bulks, pure Si/Ge square nanowires, or supperlattice Si/Ge nanolaminates and nanowires with different periodicity can be selected from the prebuilt structures. Also, users can create Si/Ge supperlattice structures with different sizes and the number of priods by their own. In addition to thermal conductivity, energies, temperature profiles, and atomic trajectories during the simulation will also be output. The second one is to run phonon relaxation time simulations on different bulk materials (e.g. Si and Ar) by spectral energy density analysis. Users can choose different temperature for their own needs. The phonon dispersion relation, relaxation time and mean free path with wave vector in 100 direction will be outputted.
Nano Heatflow

The Nano Heatflow tool allows users to explore the time evolution of kinetic and potential energy among the vibrational modes of a carbon nanotube over the course of a molecular dynamics (MD) simulation. It is possible to observe the cascade of vibrational energy through the modes of the system as a non-equilibrium population of phonons is dissipated towards thermal equilibrium, and thus gives insight into the intrinsic sources of damping and dissipation within nanoscale objects.
MiniMol
MiniMol is a minimal molecular dynamics and statics program provided with the book “Modeling Materials: Continuum, Atomistic and Multiscale Techniques” by Ellad B. Tadmor and Ronald E. Miller, Cambridge University Press, 2011.
LAMMPS for Carbon Nanostructures in MIT Atomic-Scale Modeling Toolkit

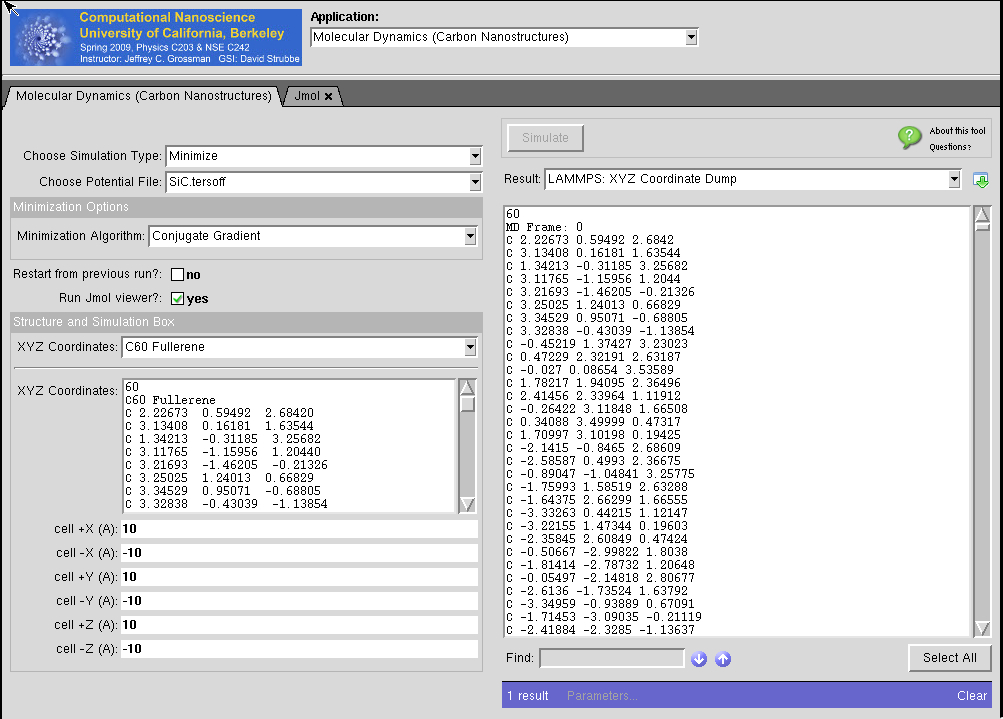
This tool is part of MIT Atomic-Scale Modeling Toolkit which serves Overview of Computational Nanoscience: a UC Berkeley Course. Powered by LAMMPS.
Lennard-Jones Potential with LAMMPS in MIT Atomic-Scale Modeling Toolkit
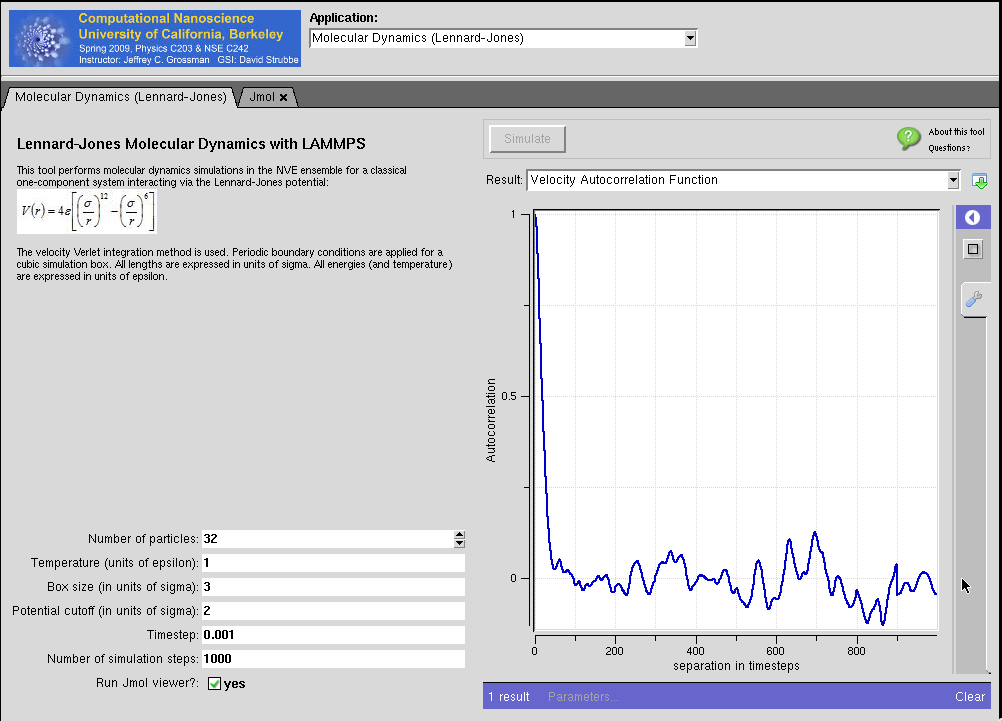 Powered by LAMMPS.
Powered by LAMMPS.
This tool is part of MIT Atomic-Scale Modeling Toolkit which serves Overview of Computational Nanoscience: a UC Berkeley Course
MIT Tools for Energy Conversion and Storage

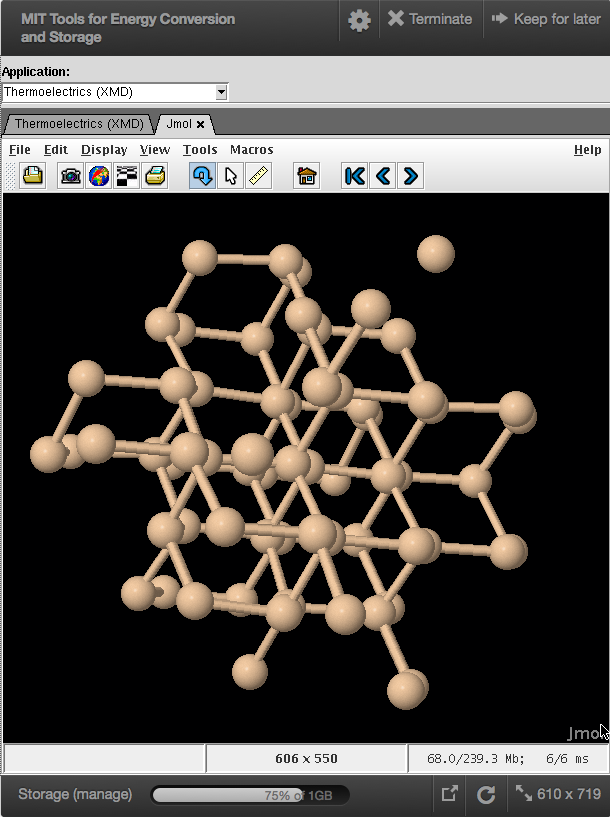
These tools allow students to focus on the atomic-scale physics and chemistry underlying four separate energy conversion and storage materials: thermoelectrics, solar fuels, solar photovoltaics, and hydrogen storage. Within each of these four different tools, the user can compute properties that are directly relevant to the key fundamental conversion and storage mechanisms.
Presentation: Polymer Synthesis and Characterization in the Cloud: a nanoHUB Classroom Experience
This talk describes an atomistic molecular dynamics laboratory on 'soft materials' that was used in a junior-level course in materials science and engineering that had over 70 participants. In this course, both molecular and continuum methods were studied and applied to relevant materials problems. The objective of the laboratory was to perform state-of-the-art molecular dynamics in series and parallel, using the Polymatic and nuSIMM computational simulation tools on nanoHUB to study polymeric materials, which are amorphous and lack long-range order or periodicity in their structure. Using these tools, students were able to set up simulations to build and visualize these materials structures and analyze the results. They developed an understanding of which method(s) can be used to solve for some important materials problems.
The assignment was designed for undergraduate students who had little or no experience with running molecular dynamics simulations. It assumed no prior knowledge of either polymatic or nuSIMM. The use of online resources (tutorials) and in person training (hands-on sessions) were demonstrated to be effective tools of engagement.
Polymatic: A Simulated Polymerization Algorithm
Polymatic is a set of codes for structure generation of amorphous polymers by a simulated polymerization algorithm. The main task of Polymatic is to perform polymerization steps within a system based on a number of defined bonding criteria. It works in conjunction with a simulation package to perform energy minimization and molecular dynamics simulations during the polymerization.
Currently, Polymatic is written to work with the Large-scale Atomic/Molecular Massively Parallel Simulator (LAMMPS) with a class I or class II force field. However, the majority of the subroutines in the code do not rely on using LAMMPS or these force field definitions, such that the code could be easily extended to work with other force fields, file types, and software packages.
Polymer Modeler

This tool provides a chain builder, with options to specify monomers, monomer arrangements (tacticity), torsion angles between monomers, system parameters such as density and temperature, as well as some prebuilt epoxy structures from current research interests. The resulting structure (constructed or prebuilt) can be used as input to LAMMPS to run molecular dynamics (MD) simulations. Many MD options are available.
nuSIMM: nanoHUB user Simulation Interface for Molecular Modeling
This tool is designed to facilitate the “synthesis in the computer” of materials and characterize them requiring only building-block atomic information.
The tool consists of three independent modules. The first module, Polymerization, assists in generating virtual polymeric materials using the Polymatic algorithm. Polymatic is implemented using the computational algorithm published by Abbott and Colina (2013) and freely available Polymatic open-source code.
The second module, Equilibration, can be used to equilibrate a low density system to a realistic density for a given temperature and pressure in an entirely predictive manner using a compression/decompression simulation procedure described by Larsen, Lin, Hart and Colina (2011).
These two steps are often used in conjunction to design nanoporous polymeric materials for which the third module, Characterization, offers structural characterization capabilities implemented using Poreblazer v2.0 developed in the Sarkisov research group and published by Sarkisov and Harrison (2011).
Crystal Viewer 2.3.4 Classic Version
Crystal Viewer is a great introductory simulation tool that allows users to create unit cells of common materials as well as carbon nanostructures that include graphene, carbon nanotubes with varying chirality, and bucky balls.
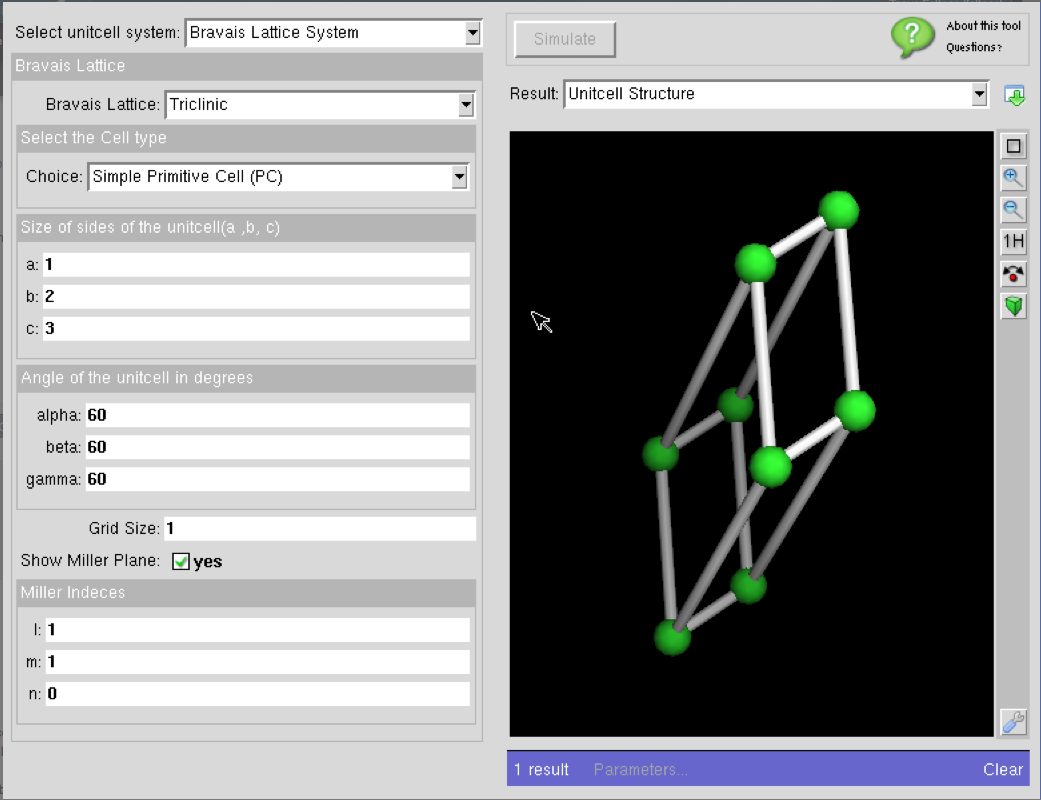
Crystal Viewer 3.0.2 All-New Version!
Crystal Viewer 3.0 is a new tool starting from scratch that is based on the old version 2.3.4 but aims to include better visualization and new features. It visualizes 14 Bravais lattices, Miller planes, and crystal structures of specific materials needed for many courses in materials science, electronics and solid state chemistry. Users can also create and view materials not included in the database. The main purpose of this educational tool is to provide insight about the crystalline structure of various materials.


Amorphous Silicon Generator
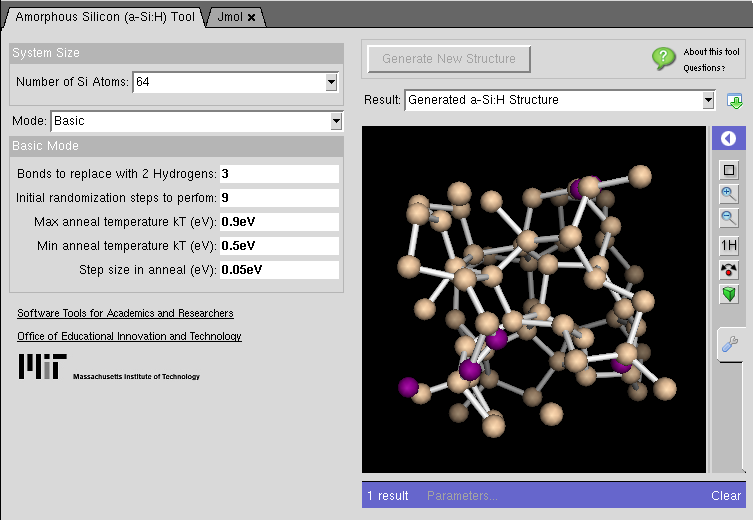
This tool generates realistic random-network models of a-Si with periodic boundary conditions.
Molecular Dynamics with Monte Carlo Simulations
See the RASPA tools, which include:
-
Void Fraction Calculator
-
Calculates the void fraction (pore volume) of nano-porous materials
-
-
Gibbs Adsorption Simulator
-
Simulates the adsorption of gases using Gibbs ensemble
-
-
Adsorption Energy Calculator
-
Calculate the total energy of adsorbates as they move around a metal organic framework
-
-
Gas Adsorption Calculator
- Simulates gas adsorption onto metal organic frameworks
-
Henry Coefficient Simulator
-
Calculate Henry's constant of several sites on a nanoporous material
-
-
Gas Diffusion Coefficient in Metal Organic Frameworks
-
Calculates gas self diffusion coefficient in metal organic frameworks
-